2025.01.27
バイオインフォマティクス・トレーニング「新科目リリースキャンペーン」

【新科目】 エッセンシャル動画視聴プラン
大学・研究所の医師や研究者、企業の研究職のみなさまの独学を支援するための新プラン「エッセンシャル動画視聴プラン」をリリースいたしました。
データ解析に役立つLinuxとRの基礎知識と活用イメージを学び、今後の研究推進に必要な技術か検討できます。
入門のためのエッセンシャル動画視聴プランから、
実践のオンデマンド実習プランへ、習熟度に合わせて学習を進められます。
<オンデマンド実習プラン>
◆すぐにコマンドを実践できる
インターネット環境さえあれば、すぐにトレーニング用解析環境をご提供。
コマンド理解とサーバ実習のセットで段階的に学びを深め
バイオインフォマティクスを実践できます。
◆現役バイオインフォマティシャンに質問できる
トレーニング内容のわからないこと、上手くいかないコマンドを
実際にデータ解析をしている現役バイオインフォマティシャンにハンズオン形式で質問できます。

キャンペーン詳細
オンデマンド実習プラン追加お申込みすれば
Linux & R エッセンシャル動画視聴プラン
実質無料!
| 対象期間 | 2025年1月27日(月)~2月28日(金) |
| キャンペーン対象 | 対象期間内にLinux & R エッセンシャル動画視聴プランをご発注された方 |
| 実施形式 | オンデマンドサイトでの受講 |
| 注意事項 | Linux & R エッセンシャル動画視聴プランのご発注日より、3ヶ月以内にオンデマンド実習プランにお申込みいただくと、1科目のみ5,000円割引(税抜)でご提供いたします。 |
Linux & R エッセンシャル動画視聴プランをクレジットカードまたは銀行振込で購入希望の方は、上記「Linux & Rご購入はこちら」ボタンよりお進みください。ご購入には、弊社学習サイト「バイオインフォマティクス・トレーニングオンデマンド」への会員登録が必要です。
▼トレーニングオンデマンドの体験動画をご視聴ください!▼
プラン詳細
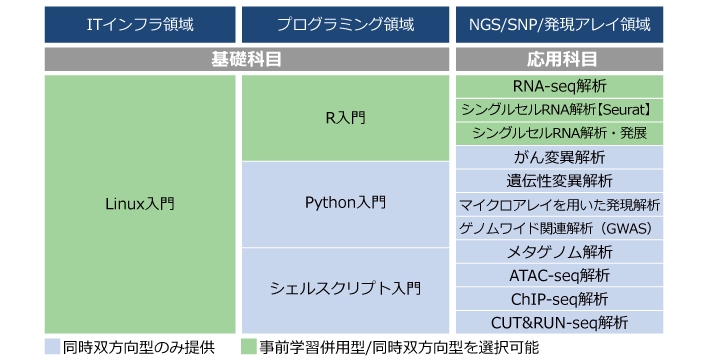
<エッセンシャル動画視聴プラン>
| 科目 | 学習内容 | 学習方式 | 価格(税抜) |
|---|---|---|---|
| Linux | ・Linuxの概要、ディレクトリ構造、絶対パスと相対パス ・ディレクトリ移動、ファイル操作・加工などの15コマンド ・ソフトウェアのインストール(PLINK) |
動画視聴 (45分) 確認テスト |
5,000円 |
| R | ・Rの概要、基本的な操作方法 ・演算、変数と代入、データ型とデータ構造 ・作図のサンプルコマンド紹介、パッケージインストール |
動画視聴 (32分) |
|
| GWAS | ・GWASの概要と論文事例、SNPアレイ解析 ・ケースコントロール解析と量的形質関連解析 ・SNPインピュテーション、PRS解析 |
動画視聴 (63分) |
5,000円 |
<オンデマンド実習プラン>
| 科目 | 学習内容 | 学習方式 | 価格(税抜) |
|---|---|---|---|
| Linux | エッセンシャル動画視聴プランの内容に加えて ・アクセス権限変更等の追加10コマンド紹介と実習 ・BLAST、Samtoolsのインストール |
・動画視聴(48分) ・サーバ実習(33問) ・ハンズオン質問会 |
60,000円 |
| R | エッセンシャル動画視聴プランの内容に加えて ・各データ構造の各種関数、基本統計量の解説と実習 ・箱ひげ図、ヒストグラム、散布図等の解説と実習 ・カテゴリ単位の処理、スクリプトファイル読込 |
・動画視聴(74分) ・サーバ実習(34問) ・ハンズオン質問会 |
60,000円 |
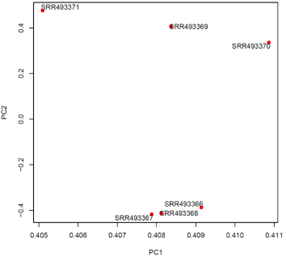
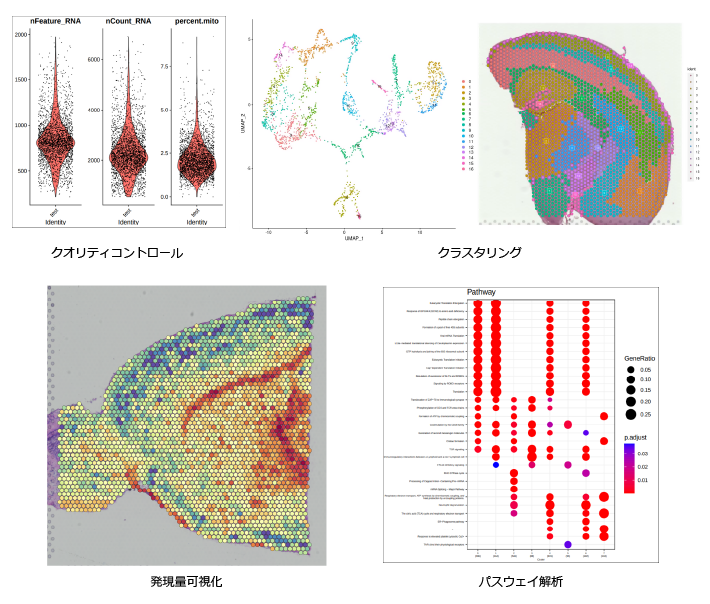
| RNA-seq | 【発現変動遺伝子の検出、二群間比較】 ・QCからGO・パスウェイエンリッチメント解析 までの実践的な実習 |
・動画視聴(74分) ・公開データ解析実習 ・ハンズオン質問会 |
80,000円 |
| シングルセル RNA-seq 【Seurat】 |
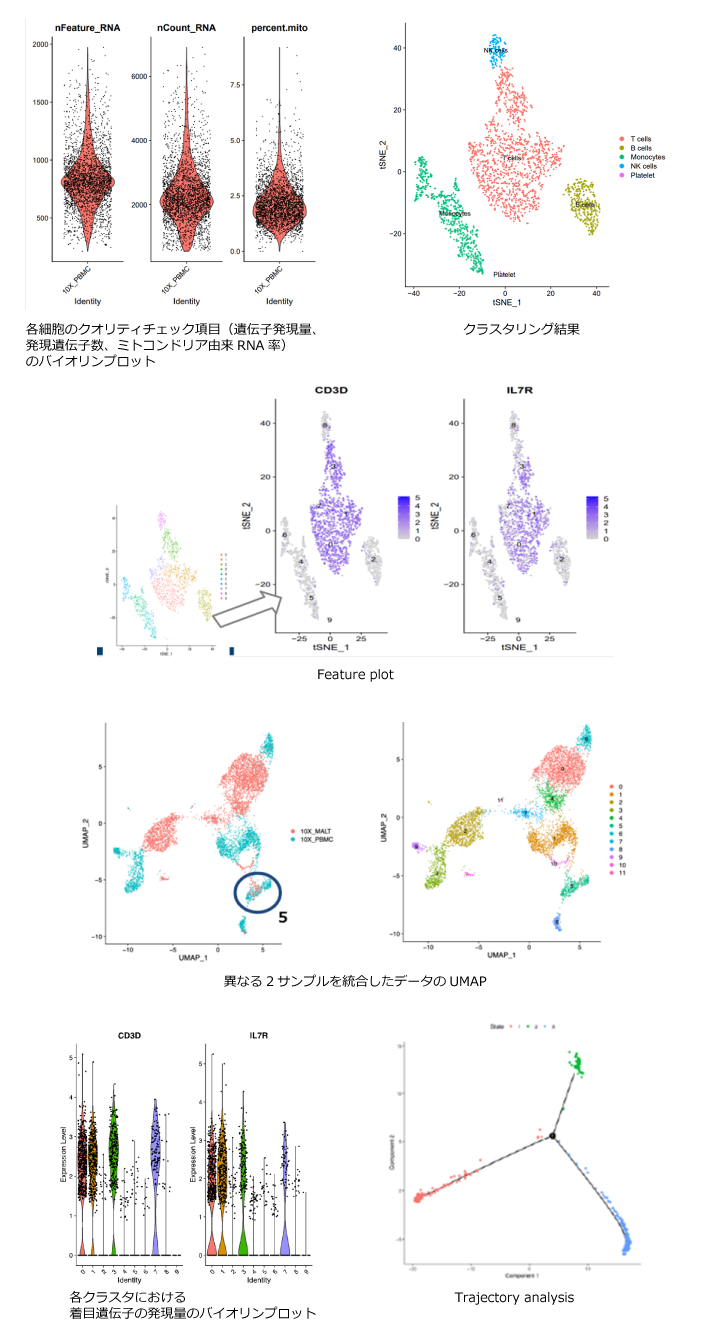
【Seuratを用いたシングルセルRNA-seq解析技術】 ・クラスタリングやマーカー遺伝子探索と セルタイプ同定などの実践的な実習 |
・動画視聴(60分) ・公開データ解析実習 ・ハンズオン質問会 |
100,000円 |
| シングルセル RNA-seq 発展 |
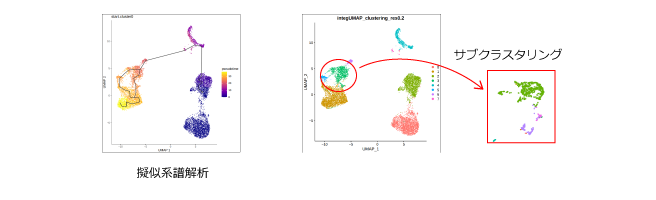
【擬似系譜解析とvelocity解析】 ・monocleを用いたtrajectory解析の解説と実習 ・velocytoを用いたRNA velocity解析の解説と実習 |
・動画視聴(58分) ・公開データ解析実習 ・ハンズオン質問会 |
100,000円 |
お客様の声
平日の昼間は忙しく、突発的なオペがあったりと学習時間を確保できなかったが、オンデマンド動画で日曜、祝日、夜間など自分の都合の良い時間に学習できた。
大学臨床研究医

整備されたサーバ上でデータ解析を繰り返し実習でき、トライ&エラーで理解度が上がってよかった。事前学習でつまづいた部分もあったが、ハンズオン質問会で解決できた。
大学臨床研究医